
| トップへ | 生物の起源 | 雑記 | 新刊紹介 | 地質年代表 | 生物分類表 |
| Genera Cellulatum | 系統解析 | リンク集 | プロフィール | 掲示板 | 系統解析トップへ |
目次 |
系統解析は今や分子生物学の基本ツールの一つになっています。しかし系統解析の具体的な方法については, あまりテキストが出回っていないのも現状です。そこでまず最初に学ぶプログラムとして,ClustalX の使い方を中心に系統解析の具体的な方法について解説を試みてみました。 より丁寧な系統解析には他の様々なプログラムの利用が推奨されますが,とりあえず系統樹を描いてみることはできると思います。
初心者向けということでなるべく詳細な解説を心がけましたが,系統樹の意味などある程度の基礎知識は前提にしていますので, そのあたりに疑問がある方はまた別の資料を探してみて下さい。
なお,本稿は Windows XP もしくは Vista の使用を前提に書かれています。従って他の OS を使用している場合にはプログラムのインストールなど, 細かい違いが生じる場合があります。
系統解析を実際に行うためには、解析用のプログラムを入手する必要があります。 多くのプログラムは無料で公開されていますが、一部には有料のプログラムもありますので、目的に応じて入手してみてください、 ここでは ClustalX というプログラムの使い方を紹介しますので、以下のサイトから最新版を入手しましょう。
ClustalX(アラインメントと近隣結合法による系統樹の作成に使います。2.0 が最新版です):
Clustal
上記のサイトに行くと、ダウンロードの欄から "Precompiled executables for Linux, Mac OS X and Windows of the most recent version along with the source code are available here" とあります。 この青文字下線部分をクリックしてください。
するとファイルのリストが表示されます。この中から、最新版の適切なファイルを選んでデスクトップなどに保存してください。 WindowsXP または Vista を使っている場合、"clustalx-2.0.4-win.msi"(バージョンは更新される可能性があります) で正常にダウンロードができました。これは ClustalX の Setup Wizard です。
さて この Setup Wizard を起動し,指示に従って ClustalX をインストールしてください。 インストール先を聞かれる他は特に選択肢はありませんので,"Next" をクリックすれば問題ありません。なお,Windows Vista の場合にはプログラムの実行を許可するか訪ねられますが,"許可" してください。これでインストールが完了します。 インストール先を変更していない場合,プログラムリストに "ClustalX2" というフォルダが生成します。ここに含まれる "ClustalX2" がプログラムの本体です。
SeaView(ClustalX で作成したアラインメントを編集するプログラムです。2.2 が最新版です。2007年10月に更新されています):
SeaView
リンク先にある該当 OS の圧縮ファイルをダウンロードし,適当なフォルダで解凍してください。 プログラムの本体は seaview.exe で,ヘルプを利用するには同じフォルダに seaview.help が必要です。
NJplot(ClustalX で作成した系統樹を表示・編集するためのプログラムです。2007 年06月に更新されています):
NJplot
リンク先にある該当 OS の圧縮ファイルをダウンロードし,適当なフォルダで解凍してください。 プログラムの本体は njplot.exe で,ヘルプを利用するには同じフォルダに njplot.help が必要です (unrooted.exe は無根系統樹の表示に使えます)。
解析を実行するプログラムが手に入りましたら、次は解析するデータが必要です。 解析したい配列は DNA の塩基配列の場合もあれば、タンパク質のアミノ酸配列の場合もあるでしょう。 また、自分で解読した配列を解析する場合もあれば、各種データベースからダウンロードした配列を比較する場合もあると思います。 ここではまず、データベースから入手した配列で系統解析を行い、 最後に自前の配列を加えて系統解析する方法についてご紹介いたします。
配列情報が登録されているデータベースは幾つか存在します。各種のデータベースにはそれぞれの特徴がありますが、 今回は NCBI(National Center for Biotechnology Information) http://www.ncbi.nlm.nih.gov/ のデータベースから配列情報を取り寄せましょう。
配列を探索するには様々な方法があります。よく利用するのは遺伝子名からの検索、 アクセッション番号(accession number)からの検索、 配列情報から類似した配列を探索する、などのケースがあると思います。遺伝子名は研究者によって表記が変わることも多いので、 遺伝子名からの検索は見逃しもあるかもしれません。その場合は、遺伝子名で代表的な配列を検索し、 その配列から類似した配列の探索を行う、など工夫が必要です。 遺伝子名やアクセッション番号からの探索には NCBI の Entrez というプログラムが利用できます。 類似した配列の探索には NCBI の BLAST というプログラムが利用できます。
Entrez を利用するには、NCBI のトップページから検索をかけます。塩基配列であればセレクトボックスから Nucleotide を、 タンパク質のアミノ酸配列であれば Protein を選択します。そして、
とある右のテキスト入力フィールドにキーワードを入力して、 ボタンをクリックすれば、 検索がスタートします。条件検索のためのオプションが幾つかありますので、以下の用例を参照して下さい (詳細は Entrez のヘルプ中にある Writing Advanced Search Statements (http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=helpentrez.section.EntrezHelp.Writing_Advanced_Sea)を参照)。
凡例
アクセッション番号が "M10098" の配列
("M10098")あるいは
("M10098[ACCN]")(後者は検索対象をアクセッション番号に限定していますが、事実上必要ありません)
アクセッション番号が "AF107923" から "AF107929" までの配列全て
("AF107923:AF107929[ACCN]")rbcL 遺伝子の配列
("rbcL[GENE]")Arabidopsis の rbcL 配列
("Arabidopsis[ORGN] AND rbcL[GENE]")("AND"、"OR"、"NOT" の論理演算子を使う場合には、大文字で入力する必要があります)
脊椎動物の cox1 配列
("vertebrates[ORGN] AND cox1[GENE]")(生物名の指定("[ORGN]" を使う)には、Entrez の Taxonomy (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Taxonomy)で使われている表記に従いましょう)
イネ(Oryza sativa)とシロイヌナズナ(Arabidopsis thaliana)の rbcL 遺伝子
(("Oryza sativa[ORGN] OR Arabidopsis thaliana[ORGN]) AND rbcL[GENE]")
BLAST を利用するには、NCBI のトップページからのリンクをたどります。あるいは、直接 NCBI BLAST (http://www.ncbi.nlm.nih.gov/BLAST/)へ行って下さい。塩基配列の検索であれば、 nucleotide BLAST へ、アミノ酸配列の検索であれば protein BLAST へ進みます。
デフォルトでは系統解析の目的に適さないことから,幾つかのオプションを設定する必要があります。
Choose Search Set:検索するデータベースの範囲を指定します。nucleotide BLAST の初期設定ではヒトのゲノムと転写産物しか検索されません。
Program Selection:検索の方法(プログラム)を選択します。
これらの設定は一度行うと記録しておくことができます。右上付近にある "Bookmark" をクリックすると,設定通りのページが開くため, この URL をコピーまたはお気に入りなどに登録しておくと便利です。
さて,設定が終わりましたら "Enter Query Sequence" のテキスト入力フィールドに、目的の配列(その配列と類似した配列を探す)を入れてください。 FASTA 形式のファイルでもかまいませんし,"参照" からファイルを読み込むこともできます。
この時、データベースなどの配列情報をコピーすると数字、スペースなどが含まれますが、これはプログラムが無視してくれるので、そのままで問題ありません。
このようにして、設定と配列の入力が完了したら、ページ下方にある"BLAST" をクリックしてください。するとページが変わり結果が表示されます。
このようにして検索した配列を見ると,以下の様な情報が得られます(各項目の詳細は,NCBI の
GenBank Sample Record
LOCUS:座位,配列の長さなど幾つかの情報が含まれます。
DEFINITION:配列に関する簡潔な説明(見出しのようなもの)です。
ACCESSION:アクセッション番号。配列の ID で,この配列を特定するための番号。論文などで引用されるのもこの番号です。
VERSION:配列のバージョン情報。配列情報が修正されるなどすると番号が変わります。
DBSOURCE:タンパク質のアミノ酸配列の場合,その元となった(塩基配列などの)データベースの ID が示されます。 ここのリンクから DNA の配列に飛べます。
KEYWORDS:キーワード。現在ではほとんど活用されません。
SOURCE:配列の由来。ORGANISM のサブ項目に由来する生物の学名が記されています。
REFERENCE:配列が出版された論文などの引用。あるいは配列の登録者の情報です。
FEATURES:配列の特徴など,様々な付帯情報。タンパク質をコードしている範囲やイントロン情報などがあります。 CDS 中の protein_id には protein データベースの当該配列へのリンクがあります。また CDS 中の translation にコードされているタンパク質のアミノ酸配列があります。
ORIGIN:配列情報。ここをコピーすることになります。
こうして得られた情報を ClustalX で読み込める形式で保存します(次項)。
NCBI などから入手した配列情報は、ClustalX で読み込める形式で保存します。読み取れるファイル形式は 7 通りありますが, ここではシンプルな Fasta 形式での保存方法を紹介します。 まず、メモ帳などのテキストエディタを開きましょう。Fasta ファイルの保存・編集に用います。
例として、(ちょっと極端な例ですが)哺乳類 3 種(カモノハシ Ornithorhynchus anatinus、 ネズミ Mus musculus、ヒト Homo sapiens)の 18S rRNA の系統解析を行う場合を考えましょう。 系統樹に根をつけるためには、調べたい生物群(内群)以外の生物を外群として用います。 ここではニワトリ Gallus gallus(もちろん哺乳類ではありません)を外群にします。
まず、ここまでで紹介した検索方法に従って 4 種の 18S rRNA の配列を検索し、 それぞれの配列のページにたどり着いたものとしましょう。とりあえず用いる配列のアクセッション番号は、 カモノハシ(AJ311679)、ネズミ (X00686)、 ヒト(M10098)、 ニワトリ(AF173612)です。
それぞれのページから配列情報を以下の形式に従ってコピーします。
>1_Ornithorhynchus_anatinus AJ311679
1 agcatatgct tgtctcaaag attaagccat gcatgtctaa gtacacacgg ccggcacagt
61 gaaactgcga atggctcatt aaatcagtta tggttccttt ggtcgctcgc tccctcctac
121 ttggataact gtggtaattc tagagctaat acatgccgac gagcgctgac ccgggcccct
(中略)
1681 aagtccctgc cctttgtaca caccgcccgt cgctactacc gattggatgg tttagtgagg
1741 tcctcggatc ggccccgccg ggggtcggcc acggcccctg gcggagcgct gagaagacgg
1801 tcgaacttga ctatctagag gaagtaaaag tcgtaacaag gtttccgtag
>2_Mus_musculus X00686
1 tacctggttg atcctgccag tagcatatgc ttgtctcaaa gattaagcca tgcatgtcta
(中略)
1861 ggatcatta
>3_Homo_sapiens M10098
1 ccgtccgtcc gtcgtcctcc tcgcttgcgg ggcgccgggc ccgtcctcga gcccccnnnn
(中略)
1921 taaaagtcgt aacaaggttt ccgtaggtga acctgcggaa ggatcatta
>4_Gallus_gallus AF173612
1 attaagccat gcatgtctaa gtacacacgg gcggtacagt gaaactgcga atggctcatt
(中略)
1681 acggccctgc cggagcgtcg agaagacggt cgaacttgac tatctagagg aagtaaa
Fasta 形式では、一行目に「>」で始まる OTU(operational taxonomic unit,操作上の分類単位)の名称と、その他の情報を、 二行目以降に遺伝子の塩基(またはアミノ酸)配列を記述することになっています。OTU の名称には全て半角英数字とアンダーバー "_" しか使わないようにしましょう。特にハイフン "-" は ClustalX では使えますが、他の多くのプログラムで使えませんので, 避ける習慣をつけておきましょう。
プログラムによって表示される OTU の文字数が異なりますので,10 文字以内で OTU を表現するか, ここで示したように頭に通し番号をつけておくと後々便利です。また一行目にスペースが存在すると,それ以降は認識されません。 そのため学名の属名と種小名の間などはアンダーバーでつなぐようにしましょう。逆に表示する必要のない情報 (ここではアクセッション番号)をスペースより後ろにメモしておくこともできます。
OTU の名称には,NCBI のデータベースにある SOURCE - ORGANISM に表示されている学名を用いると便利です。 また配列部分は ORIGIN の項目からのコピーで通常は構いません。数字、スペースなどが含まれますが,これは BLAST の場合と同様に ClastalX によって無視されるため,特に削除する必要はありません。
注:データによっては配列中にイントロン情報などのコメントが書き込まれている場合があります。 アルファベットで記述されているコメントは ClustalX に誤って認識されてしまいますので,予め削除しましょう。 あるいは,NCBI のデータシートの上部にある Display のセレクトボックス(初めは "GenBank" が選択されている)で "FASTA" を選択すると,配列情報だけが表示されます。
このようにして作成したファイルを保存します(ここでは 18S.fst とします。18S.fst http://www2.tba.t-com.ne.jp/nakada/takashi/phylogeny/data/hj2/18S.fst からダウンロードできます。メモ帳などで開いてみて下さい)。
さて,データが集まりましたらこれを並べる必要があります。例えば上記の 18S.fst ファイルに含まれる配列は, 1850 塩基対(カモノハシ),1869 塩基対(ネズミ),1969 塩基対(ヒト),1737 塩基対(ニワトリ) とそれぞれ長さが違います。これは解読されている領域そのものが異なっているのかもしれませんし, 進化の過程で挿入・欠失が蓄積して遺伝子の長さそのものが変わったのかもしれません。
しかし系統解析を行うためには相同な領域同士を比較する必要があります。そのために配列間で相同な領域を推定し, 相同な座位が縦に並ぶように挿入/欠失の位置を推定する操作を「アラインメント」(alignment)と言います。 アラインメントには ClustalX というプログラムを用いると便利ですので,このプログラムの使用法について紹介します。
ClustalX を開きます。プログラム一覧から ClustalX2 を開きます。次に File → Load Sequences から作成した Fasta 形式のファイルを読み込みます。
注:ClustalX 2.0 では,ファイル名またはファイルのパス(ファイルを右クリックしてプロパティを開くと, "場所" として表示されます)に日本語が含まれていると読み込めません。特に Windows XP では,デスクトップに置いたファイル, あるいはデスクトップに置いたフォルダ内部のファイルは読み込めません(Windows Vista の場合は問題ありません)。 ファイル名,フォルダ名を日本語に直すか,新たにアルファベット表記のフォルダを作成して読み込みたいファイルを移して下さい。
画面に表示された配列を見ると,配列がそろっていないことが多いかと思います。上述の 18S.fst を読み込んだ場合, 配列はばらばらになっています(図 1)。
 |
| 図 1 |
Alignment → Do Complete Alignment としてアラインメントを実行します。ファイルの保存先が聞かれますが, 表示されている保存先は読み込んだファイル(18S.fst)と同じディレクトリで,ファイルの名前は,拡張子が .dnd と .aln に変更されたものです。これは必要に併せて変更してください。同じ名前のファイルが既に存在する場合でも, 警告無く上書きされますので注意してください(特にアラインメントをやり直す場合など)。
しばらく待つと(数十秒〜数十分)画面が更新され,アラインメントが済んだ状態が表示されます。 18S.fst の場合はヒトの配列の 5' 末端が長く,100 塩基ほどはヒトの配列だけです。 ヒト以外の 5' 末端の配列を例示すると,図 2 の様になっています。
注:M10098(ヒトの配列)の FEATURES を見ると,18S ribosomal RNA は 102 番目の塩基からだと分かります。
 |
| 図 2 |
この例の場合,18S.aln(18S.aln http://www2.tba.t-com.ne.jp/nakada/takashi/phylogeny/data/hj2/18S.aln からダウンロードできます)と 18S.dnd という二つのファイルが生成します。前者がアラインメントのファイルです。 これは ClustalX や テキストエディタで開くことができます。後者はアラインメントの過程で作られる暫定的な系統樹で, アラインメントに基づいていないため,これを系統樹として利用しないように注意してください。
アラインメントを見ると,似た配列が縦に並ぶように左端がずらされていることがわかります。しかし, 場所によってはアラインメントに問題があることもあります。そのような場合は,手動で再編集する必要があります。
例えば図 3 の様なアラインメントの間違いがあります。
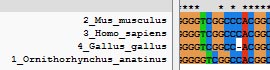 |
| 図 3 |
"TC" という並びは明らかに全ての配列で相同ですが,種によってずれていることが分かります。これは ClustalX がなるべくギャップ("-" で表示される)を挿入しないように並べるために起こる問題で, 今回のように露骨な場合には手動で修正した方が良いでしょう。
メモ帳などテキストエディタを用いて直すこともできますし,アラインメントを編集するプログラムも様々なものが公開されています
(本サイトでは SeaView の使い方を解説しています。SeaView を用いたアラインメント編集
この部分にはギャップを一つ挿入し,図 4 のように修正するとよいでしょう(この場合は見た目でアラインメントが可能ですが, 正確には 18S rRNA の二次構造のモデルなどを参考に,厳密に判断する必要があります)。
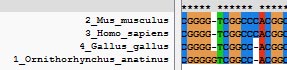 |
| 図 4 |
一方で,配列間で置換が多すぎてアラインメントができない領域も存在します。二次構造を参考にすれば可能かもしれませんが, 図 5 の様な例ではどの生物のどの部分が相同なのか判断できないため,系統解析には用いない方がよいでしょう。
 |
| 図 5 |
先と同様にこの領域を削除して図 6 のように編集します。
 |
| 図 6 |
このようにして編集し,相同な配列のみを残したファイルを保存します
(ここでは 18Sed.aln とします。18Sed.aln
作成したアラインメントに自分が解析した配列などを後から追加することもできます。ここではオポッサムの一種 (Didelphis virginiana)の 18S rRNA の配列 (AJ311677) を後から追加して,その配列のアラインメントだけをやり直すことを考えましょう。
データの収集と Fasta ファイルの作成で説明したように,オポッサムの配列を別の Fasta
形式のファイルに保存します(ここでは 18Sop.fst とします。18Sop.fst
ClustalX で編集後のアラインメントファイルを開きます(例:18Sed.aln)。
File → Append Sequences として,追加したい配列を含んだファイルを読み込みます(18Sop.fst)。
新たに追加した配列の OTU の名称をクリックします。するとその OTU が水色に選択されます。
この状態で Alignment → Realign Selected Sequences とすると,この配列だけを再度アラインメントすることができます。 Do Complete Alignment の場合と同様にファイルの保存先を設定します(ファイル名の本体は最初に読み込んだファイルになっています)。 ここで OK をクリックするとアラインメントが始まります。
配列のアラインメント作成のときと同様に配列を編集し,アラインメントを整えます
(ここでは 18Soped.aln とします。18Soped.aln
いよいよ系統樹の作成です。作成したアラインメントはファイル形式を変換することで, 様々なプログラムを用いた系統解析に利用できます。ClustalX でも近隣結合(Neighbor Joining:NJ)法と平均距離 (Unweighted Pair-Group Method using Arithmetic averages:UPGMA)法を用いた系統解析が利用できますので,その方法を紹介します。
ClustalX に戻り,編集したファイルを File → Load Sequences から読み込みます。この画面で, 編集したアラインメントに問題がないか再確認しておくといいでしょう。
Trees からオプションを選びます。
Draw Tree:Clustering Algorithm で設定した方法で系統樹を描きます。統計的な支持率の計算はしませんが, その分早く解析が終わります。下記のオプションで設定を決めてから選択しましょう。生成するファイルの拡張子は **.ph です。
Bootstrap N-J Tree:ブートストラップ値の付いた NJ 系統樹を描きます。Clustering Algorithm で "NJ" を設定したときにのみ選択可能です。ブートストラップ値は系統樹の枝の信頼性を表す統計値で, ランダムに改変したアラインメントを大量に生成し,それぞれのアラインメントから作られる系統樹の内いくつが問題の枝 (単系統群)を含んでいるかを数えます。従ってブートストラップの回数が増えれば増えるほど時間がかかります。 通常の NJ 系統樹の場合と同様にオプションを設定しましょう。生成するファイルの拡張子は **.phb です。
Exclude Positions with Gaps:系統解析の際に,アラインメントの中にあるギャップを含んだ座位を除くオプションです。 ギャップが解析に悪影響を与えると考えられる場合にはこのオプションにチェックを入れておく必要があるでしょう。 ただしギャップを排除した場合に,使える座位数が少なくなりすぎる場合もあるので,注意が必要です。
Correct for Multiple Substitutions:系統解析の際に,配列間の距離を補正するオプションです。 配列間で塩基置換が多い場合,同じ座位に 2 回以上の置換が起きることがあります。これを補正して, 配列間の距離を計算する計算式は幾つかありますが,塩基配列の場合には Kimura の二変数法(Kimura, 1980), アミノ酸配列の場合には Kimura (1983) による補正距離が用いられます。ごくごく近い系統間の比較を行う場合を除いて, チェックを入れておくとよいでしょう。
Output Format Options:出力されるファイルの種類や保存形式を指定するオプションです。 系統樹を見るためのプログラムに応じて選択しましょう。今回紹介している NJplot では初期設定のままで構いません (Output Files は Phylip format tree,Bootstrap labels on: では Branch)。この他によく用いられるプログラムに TreeView がありますが,これを用いる場合にはOutput Files に Nexus format tree を,Bootstrap labels on: では Node を設定して下さい。 なお,複数のファイル形式を同時に選択することも出来ますが,"Phylip distance matrix" と "% identity matrix" を選択しても, ブートストラップ解析には利用できません("Draw tree" の場合のみ利用できます)。
Clustering Algorithm:系統解析の方法を選択します。UPGMA 法と NJ 法が選択可能で, 初期設定では NJ 法が選択されています。なお,UPGMA 法を選択した場合にはブートストラップ値を計算できません。
ここでは Correct for Multiple Substitutions にチェックを入れて,Bootstrap N-J Tree を実行することにしましょう。
Bootstrap N-J Tree を選択すると,3 つの設定項目が表示されます(Draw Tree の場合にはファイルの場所/名前のみ) 。
Random number generator seed [1-1000]:乱数の種を設定します(初期値は 111)。ブートストラップのために, ランダムにアラインメントを作成するための乱数を決める値です。この値が同じであれば結果は同じになりますし, この値を変えれば異なる乱数に基づいてアラインメントを作成します。通常は変更する必要はありません。 変更したい事情がある場合には,1-1000 までの値を入力します。
Number of bootstrap trials [1-10000]:ブートストラップの回数を設定します(初期値は 1000)。 1000 回にするのが一般的ですが,アラインメントのデータが巨大であったり,コンピュータの性能が低い, などやむを得ない場合には 100 回や 500 回にすることもあるかもしれません。増やす意味はあまりないと思います。
Save Phylip tree as:ファイルの保存場所と名称を指定します。初期値は最後に読み込んだファイルの拡張子を **.phb (Draw Tree の場合には **.ph)にしたものです。必要に併せて変更して下さい。 なお,同じ名称のファイルが存在する場合には,警告抜きで上書きされますので注意して下さい。また,Output Format Option で複数のファイル形式を選択している場合には,それぞれに対応して "Save ****** tree as" と表示されます。 ただし "Phylip distance matrix" や "% identity matrix" を選択しても,ブートストラップ解析の場合には表示されません。
ここでは初期値のままで解析を行います。データのサイズによって必要時間は数秒から数時間,と異なります。 ブートストラップ値を求める場合には,おおよそ Draw Tree での所要時間×ブートストラップの回数だけの時間がかかると思われます。
系統樹のファイルが出力されたら(ここでは 18Soped.phb
NJplot(njplot.exe)を開きます。
File → Open として,系統樹のファイル(18Soped.phb など)を開きます。
ブートストラップ値を伴わず,適当な外群に基づいて描かれた系統樹が表示されます。
まず系統樹の見栄えを編集します。Operation のチェック項目が 4 つあります。
Full tree:系統樹の全体を表示させます。初めは全体が表示されていますが, 下記の 3 つのいずれかの選択中に用います。
New outgroup:外群を選択します。外群は初め適当に選択されていますので,自分で適切な外群を設定しましょう。 外群となる生物,あるいは系統群のところにある "#" をクリックすると,その枝を外群とした系統樹が表示されます。
Swap nodes:枝の上下を入れ替えます。OTU の配置には生物学的な意味はありませんが, 表示の上下を入れ替えた方が見やすくなることはしばしばあります。"#" をクリックすることでその上下の枝が入れ替わります。 あくまで見やすさのための機能なので,気にならない人には不要かもしれませんが,意外に便利です。
Subtree:系統樹の一部のみを表示します。"#" の右側だけが切り出されて表示されます。系統樹が大きすぎるなどして, 一部だけを見せたい場合に利用できます。一度部分系統樹を表示したあとでも,すぐ下あたりにある Subtree up のボタンをクリックすると一つ左(上位)の枝までが表示されます。また,Full tree でもとに戻ります。
この中で,外群の設定(New outgroup)の設定は最も重要ですので,外群があっている場合を除き,必ず設定し直しましょう。 最後に Full tree(または Subtree)を選び,次に進みます。
ブートストラップ値や枝長の表示を設定します。Display のチェック項目が 2 つあります。
Branch lengths:これにチェックを入れると,各枝の長さ(サイトあたりの塩基置換数)が数字で表示されます。 あまりこの値を用いることはありませんが,必要になることもあるかもしれません。
Bootstrap values:これにチェックを入れると,ブートストラップ値(絶対値)が表示されます。 1000 回ブートストラップを行った場合,1000 回中にその枝が出現した回数が表示されます。 通常は表示を実行したブートストラップの回数で割った値の百分率を,ブートストラップ確率として示します。 小数点以下は切り捨てましょう。
この他,Zoom で系統樹の拡大や縮小を行うこともできます。また,ツールバーには OTU 名の検索や, フォントサイズや紙のサイズなどの設定など周辺的なオプションがあります。
編集した後の系統樹は,File → Save Rooted Tree または Save Unrooted Tree で Phylip format(**_root.phb または **_noroot.phb) に保存することができます。前者では "New outgroup" で設定した外群選択を保存します。
注:File → Save as PDF という PDF 形式に保存する機能は Windows XP や Windows Vista では利用できません。
樹形をイラストの編集に持ち込む場合には,系統樹を普通にコピーするか(Edit → Copy)画面のコピー(windows の場合は Alt + Prt Sc などのキー) を用いるといいでしょう。後は,イラストを編集できるソフトを用いて,OTU の名前やブートストラップ値の表記などを手直しして完成です。 図 7 は例に用いた配列の系統樹です。ブートストラップ値は % で表記しています。オポッサム(D. virginiana)はカモノハシ (O. anatinus)よりもヒト(H. sapiens)やネズミ(M. musculus)に近縁であることがわかっていますが、 18S rRNA の系統樹では誤った結果が出ています。しかしカモノハシとオポッサムの近縁性を支持するブートストラップ値は,79% と低めになっています。
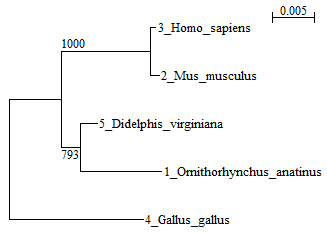 |
| 図 7 |
ここまで紹介したのは簡単な系統樹の作成法です。投稿用の論文に載せる系統樹を作る場合には,より丁寧に解析された, 複数の系統樹を比較することが望ましいと思います。それでも尚,ClustalX を用いて描いた系統樹を論文に利用したい場合, 引用するべき論文を紹介しておきます。文脈によってはこの他の論文も引用することはあるかもしれませんが, とりあえずの参考にしてみて下さい。
Larkin, M. A. et al. ClustalW and Clustal X version 2.0. Bioinformatics 23, 2947-2948 (2007).
ClustalX 2.0 のプログラムの解説をした論文です。ClustalX 2.0 を用いてアラインメント,系統樹の作成を行った場合に引用しましょう。
Galtier, N., Gouy, M. & Gautier, C. SEAVIEW and PHYLO_WIN: Two graphic tools for sequence alignment and molecular
phylogeny. Comput. Appl. Biosci. 12, 543-548 (1996).
SeaView などのプログラムを解説した論文です。SeaView を引用したい場合,この論文を。
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees.
Mol. Biol. Evol. 4, 406-425 (1987).
NJ 法を最初に発表した論文です。NJ 法を用いた場合に引用しましょう。
Sokal, R. R. & Michener, C. D. A statistical method for evaluating systematic relationships.
Univ. Kansas Sci. Bull. 38, 1409-1438 (1958).
UPGMA 法のアルゴリズムを導入した論文です。UPGMA 法を用いた場合に引用しましょう。
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of
nucleotide sequences. J. Mol. Evol. 16, 111-120 (1980).
Kimura の二変数法の典拠。塩基配列から系統樹を描く際に,Correct for Multiple Substitutions を行った場合に引用しましょう。
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge University Press, Cambridge, 1983).
分子進化の中立説の本。アミノ酸配列から系統樹を描く際に,Correct for Multiple Substitutions
を行った場合に引用しましょう。
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39,
783-791 (1985).
ブートストラップ法を系統解析に応用することを提唱した論文。系統樹を描く際にブートストラップを行った場合に引用しましょう。
Perrière, G. & Gouy, M. WWW-Query: An on-line retrieval system for biological sequence banks. Biochimie
78, 364-369 (1996).
NJplot などのプログラムを解説した論文です。NJplot を引用したい場合,この論文を。
Hall, B. G. Phylogenetic Trees Made Easy: a How-To Manual 2nd ed. (Sinauer Associates, Sunderland, 2004).
根井正利 および クマー, S. 分子進化と分子系統学 (培風館, 東京, 2006).
埼玉大学総合科学分析支援センタ−:
科学教養セミナー 第9回サマースクール
他、プログラムのダウンロード元、引用先についてはページ上部のリンク先と、各プログラムの解説などを参照して下さい。
| トップへ | 生物の起源 | 雑記 | 新刊紹介 | 地質年代表 | 生物分類表 |
| Genera Cellulatum | 系統解析 | リンク集 | プロフィール | 掲示板 | 系統解析トップへ |